- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT The storage of lipids as energy in adipose tissue (AT) has been conserved over the course of evolution. However, substantial differences in ATs physiological activities were
reported among species. Hence, establishing the mechanisms shaping evolutionarily divergence in ATs transcriptomes could provide a deeper understanding of AT regulation and its roles in
obesity-related diseases. While previous studies performed anatomical, physiological and morphological comparisons between ATs across different species, little is currently understood at the
molecular phenotypic levels. Here, we characterized transcriptional and lipidomic profiles of available subcutaneous and visceral ATs samples across 15 vertebrate species, spanning more
than 300 million years of evolution, including placental mammals, birds and reptiles. We provide detailed descriptions of the datasets produced in this study and report gene expression and
lipid profiles across samples. We demonstrate these data are robust and reveal the AT transcriptome and lipidome vary greater among species than within the same species. These datasets may
serve as a resource for future studies on the functional differences among ATs in vertebrate species. SIMILAR CONTENT BEING VIEWED BY OTHERS IDENTIFICATION OF DISTINCT TRANSCRIPTOME
SIGNATURES OF HUMAN ADIPOSE TISSUE FROM FIFTEEN DEPOTS Article Open access 13 July 2020 COMPREHENSIVE EXPRESSION ANALYSIS OF HORMONE-LIKE SUBSTANCES IN THE SUBCUTANEOUS ADIPOSE TISSUE OF THE
COMMON BOTTLENOSE DOLPHIN _TURSIOPS TRUNCATUS_ Article Open access 31 May 2024 SNRNA-SEQ REVEALS A SUBPOPULATION OF ADIPOCYTES THAT REGULATES THERMOGENESIS Article 28 October 2020
BACKGROUND & SUMMARY The adipose tissue (AT) is one of the most important organs ensuring energy and metabolic homeostasis in vertebrates1. In recent years, AT has gained sustained
scientific attention due to the significant increase in global rates of obesity and metabolic disorders in human populations, in particular type II diabetes and cardiovascular disease.
Recent studies showed that the AT is a remarkably complex organ playing important roles in energy storage, pathophysiology, and a variety of biological processes, such as controlling blood
pressure, reproduction and host defense2,3. The AT is distributed across the body4 and can be divided into the intra-abdominal visceral AT (VAT) – located around the omentum, intestines,
gonad, pericardium and perirenal areas, and the subcutaneous AT (SAT) – located in the buttocks, thighs, and abdomen. ATs from different locations have distinct properties, including
different metabolic functions, structural roles or association with diseases5,6,7,8. A previous study suggested that the root of AT complexity emerged during the course of evolution9 due to
differences in AT properties between species10, which can be evaluated by performing cross-species comparisons. A recent comparison between humans and mice identified different proportions
of a subpopulation of adipocytes regulating thermogenesis between the two species11, partly explaining observed differences in thermogenic activity. Moreover, well-documented comparative
transcriptome analyses across phylogenies can advance translational medicine by identifying novel therapeutic targets12. For example, a former study found that miR-26a, a microRNA involved
in cardiomyocyte proliferation, is down-regulated in injured zebrafish hearts but remains constant in mice12. The knockdown of miR-26a in post-natal mice hearts prolonged the proliferative
window of cardiomyocytes, indicating this miRNA could be a therapeutic target for treating heart damaged12. Accordingly, evaluating AT changes at the molecular-level across species will
enhance our understanding of the function and genetic basis of AT and its association with different diseases. Transcriptional information is important to elucidate AT phenotypes and
function, but so far most studies only focused on AT comparisons between humans and rodents13,14,15. Importantly, large-scale comparative AT transcriptomic analysis across various
distantly-related species and multiple anatomical locations is necessary to fully understand AT transcriptomic evolution. To this purpose, we performed a comparative transcriptomic analysis
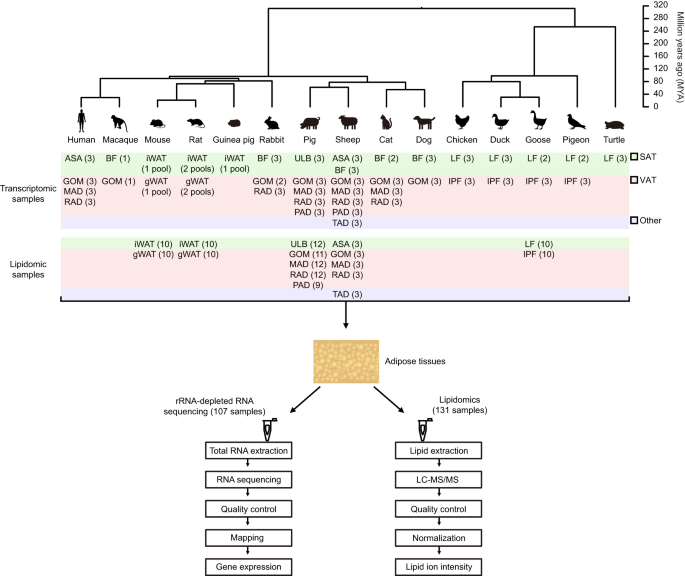
of available subcutaneous and/or visceral AT across 15 vertebrate species and locations (from 1 to 7 per species) (Fig. 1, Supplementary Table 1), including 10 mammals (primates: human
[_Homo sapiens_] and macaque [_Macaca mulatta_]; rodents: mouse [_Mus musculus_], rat [_Rattus norvegicus_], and guinea pig [_Cavia porcellus_]; lagomorphs: rabbit [_Oryctolagus cuniculus_];
artiodactyls: pig [_Sus scrofa_] and sheep [_Ovis aries_]; and carnivores: cat [_Felis catus_] and dog [_Canis lupus familiaris_]), 4 birds (galliformes: chicken [_Gallus gallus_];
anseriformes: duck [_Anas platyrhynchos_] and goose [_Anser anser_]; and columbiformes: pigeon [_Columba livia_]), and one reptile (testudines: turtle [_Pelodiscus sinensis_]) as the
outgroup. We generated a total of 59 paired-end rRNA-depleted RNA-seq libraries, and analyzed in combination with 48 libraries that were published previously16,17,18,19,20,21, totaling 107
libraries (Fig. 1, Supplementary Table 1). The lipidome composition of the ATs can impact multiple aspects of energy homeostasis, such as glucose and lipid metabolism, substrate availability
and energy expenditure22,23,24,25. Accordingly, understanding the differences in lipid composition between ATs is essential for studying their specialized functions and exploring the
potential mechanisms leading to AT heterogeneities. Lipidomics has been successfully applied previously for clarifying lipid profile changes of ATs after various treatments (such as
endurance exercise training26, cold exposure27 and high-fat diet28) or between different anatomical locations29. However, changes across species remain poorly understood. To gain further
insights into the metabolic changes that occurred during AT evolution, we performed untargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of the cellular lipidome of
131 SAT and VAT samples across five representative species, including four mammals (mouse, rat, pig, sheep) and a bird (goose) (Fig. 1, Supplementary Table 2). Altogether, these datasets
provide a valuable resource for the study of AT genetic and metabolic diversity across species and anatomical locations, and an unprecedented opportunity to analyze molecular changes during
AT evolution. METHODS ETHICS STATEMENT All research involving animals in this study were conducted in accordance with the guidelines of the Administration of Affairs Concerning Experimental
Animals established by the Ministry of Science and Technology of China. The experiments were approved by the Institutional Animal Care and Use Committee of the College of Animal Science and
Technology, Sichuan Agricultural University, Sichuan, China under permit No. DKY-2019102006. SAMPLE COLLECTION AND RNA LIBRARY PREPARATION The 107 adipose tissue samples (Fig. 1) were
obtained from various sources, including 48 previously published samples16,17,18,19,20,21 (Supplementary Table 1), and collected from 1–20 healthy adults from each of the 15 species. All
species are represented by female individuals, except for the humans (all individuals are males). Animal health was evaluated daily by a specialized laboratory animal technician (and
veterinarian, if necessary). The anatomical locations of these adipose tissues are: (1) Subcutaneous adipose tissues: Abdominal subcutaneous adipose (ASA) of humans and sheep were taken from
the abdominal wall, specifically from the area adjacent to the umbilicus in humans and the ventral lower abdominal area close to the mid-line in sheep; Back fat (BF) from macaque, rabbits,
pigs, sheep, cats and dogs refers to the subcutaneous adipose tissue located in the mid-lower part of the back, near the dorsum midline. We note that, since the back fat of adult pigs
usually has two distinct layers, we only collected the upper layer (close to skin, annotated as ULB in Fig. 1); Inguinal white adipose tissue (iWAT) of mice, rats and guinea pigs refers to
the subcutaneous adipose tissue obtained from the inguinal region of both hindlimbs; Leg fat (LF) of chickens, ducks, geese, pigeons and turtles is the subcutaneous adipose tissue collected
from the leg area, specifically from the upper portion of the legs (proximal to the pelvic bone) in chickens, ducks, geese and pigeons, and the dorsal base of the hind limbs in turtles. (2)
Visceral adipose tissues: Greater omentum (GOM) from humans, macaque, rabbits, pigs, sheep, cats and dogs refers to an apron like structure that extends from the greater curvature of the
stomach and the proximal part of the duodenum; Mesenteric adipose (MAD) of humans, pigs, sheep and cats was collected from the mesentery adjacent to the intestines; Retroperitoneal adipose
(RAD) of humans, rabbits, pigs, sheep and cats is located behind the kidney, and does not contain fat surrounding the kidney; Gonadal white adipose tissue (gWAT) from the mice and rats (all
individuals are females) is located around the ovary; Pericardial adipose (PAD) of pigs and sheep is located between the epicardium and parietal pericardium; Intraperitoneal fat (IPF) of
chickens, ducks, geese and pigeons refers to the adipose tissue attached to the gizzard. (3) Other type adipose tissues: Tail adipose (TAD) of sheep was taken from the tail base (5–7 caudal
vertebrae). The animals generated in this study for RNA-seq were commercially obtained (the mice, rats, rabbits, sheep, cats and dogs were obtained from the Chengdu dossy experimental
animals Co., Ltd, Chengdu, China; macaque and pigs were purchased from the Sichuan Hengshu Bio-Technology Co., Ltd, Yibin, China; pigeons were obtained from the Fengmao Meat Pigeon Breeding
Professional Cooperative In Fucheng District of Mianyang City, Mianyang, China; chickens were purchased from the poultry breeding farm of Sichuan Agricultural University, Ya’an, China; ducks
and geese were obtained from the waterfowl breeding center of Sichuan Agricultural University, Ya’an, China; and turtles were purchased from the Sichuan Longhu Lohas Turle Industry Ltd,
Meishan, China), and humanely sacrificed to ameliorate suffering, in accordance with the national regulations for the care and use of research animals. All samples were immediately
homogenized in liquid nitrogen and stored at −80 °C until RNA extraction was performed. Total RNAs were extracted using the standard protocol of the RNeasy Mini Kit (Qiagen, Valencia, CA,
USA). RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Only samples with high quality RNA (RNA integrity [RIN] score > 7) were used for sequencing. The
RNA-seq libraries were then generated using an rRNA depletion method18. All libraries were sequenced on a HiSeq X Ten platform (Illumina) using paired-end sequencing reads of 150 bp in
length. RNA-SEQ DATA PROCESSING Paired-end reads were aligned to the corresponding reference genomes (human: GRCh38.p13, macaque: Mmul_8.0.1, mouse: GRCm38.p6, rat: Rnor_6.0, guinea pig:
Cavpor3.0, rabbit: OryCun2.0, pig: Sscrofa11.1, sheep: Oar_v3.1, cat: Felis_catus_9.0, dog: CanFam3.1, chicken: GRCg6a, duck: CAU_duck1.0, and turtle: PelSin_1.0; goose30 and pigeon
[GenBank: WOFI01000000] reference genomes were housely constructed) using the STAR alignment tool (2.5.3a)31 with default parameters. Transcriptional abundance of protein coding genes (PCGs)
was estimated as transcripts per million (TPM) using the high-speed transcript quantification tool Kallisto (0.44.0)32. The gene annotation file was downloaded from Ensembl (Release 89). We
considered a gene as transcribed if its expression value was > 0.5 TPM in all replicates of at least one AT. IDENTIFICATION OF SINGLE-COPY ORTHOLOGOUS GENES The single-copy orthologous
PCG families in the 15 species were identified according to the Ensembl-recommended protocol (http://asia.ensembl.org/info/genome/compara/homology_method.html). Briefly, for each species, we
extracted the longest translation of each PCG, and subsequently performed all-against-all blast between self and non-self-species. Based on the blast results, we generated a sparse graph
and extracted the clusters using _hclust_sg_. The clusters with over 400 PCGs were recursively split into smaller ones until all clusters contained fewer than 400 PCGs. For each cluster, we
performed multiple alignments of protein-coding sequences and back-translated to coding sequence alignments. Finally, we constructed a phylogenetic tree and identified single-copy
orthologous gene families. UNTARGETED LIPIDOMICS SAMPLE PREPARATION AND LC-MS/MS ANALYSIS We chose the SAT and VAT samples of four mammals (mouse, rat, pig, sheep) and a bird (goose) to
investigate AT lipidome divergence across species (Supplementary Table 2). All of the animals are healthy adult females, and they were obtained in the same way as the RNA-seq study mentioned
earlier. The specific sample information are as follows: (1) subcutaneous adipose tissue: iWAT from mice and rats, ULB from pigs, ASA from sheep, and LF from geese. (2) visceral adipose
tissue: gWAT from mice and rats, GOM, MAD, RAD and PAD from pigs, GOM, MAD and RAD from sheep, and IPF from geese. (3) other type adipose tissue: TAD from sheep. The anatomical locations of
these adipose tissues were detailed in ‘_SAMPLE COLLECTION AND RNA LIBRARY PREPARATION’_ above. Total lipid extraction was performed according to a previously described method33 with some
modifications. Briefly, 25 mg of adipose tissue was added in 800 μL precooled dichloromethane/methanol (3:1, v/v). After this, 10 μL of internal lipid standards (SPLASH Lipidomix, Avanti
Polar Lipids, USA) and two steel balls were added. The mixture was homogenized using a TissueLyser for 4 min at 55 Hz. After incubation for 2 h at −20 °C, the mixture was centrifuged at
30,000 × g for 20 min at 4 °C. The 500 μL of supernatant was transferred to a new centrifuge tube and dried using a freeze-dryer. After this, 500 μL of reconstitution solvent
(isopropanol/acetonitrile/water = 2:1:1, v/v/v) was added and centrifuged at 25,000 × g for 20 min at 4 °C. Next, 100 μL of supernatant was transferred to a new centrifuge tube containing
500 μL of reconstitution solvent. 80 μL of each sample was then transferred to the LC/MS vial. In addition, to monitor system stability, a quality control (QC) sample was prepared by
combining the same volume of all experimental samples. The lipid extracts were analyzed using ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) (Waters,
Manchester, UK). Each sample (5 μL) was injected onto a CSH C18 column (2.1 × 100 mm, 1.7 μm, Waters), which was kept at 55 °C with an elution rate of 0.4 mL/min. The mobile phase A was ACN:
H2O (60:40 v/v) and the mobile phase B, was IPA: ACN (90:10 v/v), both containing 0.1% formic acid and 10 mM ammonium formate. The gradient elution conditions were: 0–2 min, 40–43% phase B;
2.1–7 min, 50–54% phase B; 7.1–13 min, 70–99% phase B; 13.1–15 min, 40% phase B. The Xevo G2-XS QTOF mass spectrometer (Waters, UK) was used to detect the metabolites eluted from the
chromatographic column in both positive and negative ion modes. The positive or negative ionization mode parameters were as follows: capillary voltage 3 (pos)/2 (neg) kV; cone voltage 40 V;
source temperature 120 °C; deconvolution temperature 450 (pos)/350(neg) °C. The mass spectrometry data were acquired in Continuum MSE mode. The scanned _m/z_ range of MS signal varied
between 100 to 2000 Dalton in the positive ion mode, and 50 to 2000 Dalton in the negative ion mode. The survey scan time was 0.2 s. Based on the precursor ion intensity, the top 3 ions were
selected for MS2 analysis. For the MS/MS detection, the precursors were fragmented using 19–45 eV with a scan time of 0.2 s. The leucine enkephalin signal was measured every 3 s during
acquisition to calibrate the mass accuracy. LC-MS/MS DATA PROCESSING The raw data from the mass spectrometer was imported into the commercial software Progenesis QI (version 2.2, Nonlinear
dynamics, http://www.nonlinear.com/progenesis/qi) for peak detection and alignment. After this, the peak intensity data were further processed according to a previously described metaX
pipeline34 with slight modifications. Firstly, we retained the high-quality ions present in > 50% of QC samples and > 20% of the replicates of at least one AT. The missing values were
imputed using the k-nearest neighbor (KNN) method35,36. Next, the probabilistic quotient normalization (PQN) method37 was performed for data normalization and the QC-robust spline batch
correction (QC-RSC) method38 applied to correct for batch effects. Finally, the ions with a relative standard deviation (RSD) > 30% in QC samples were removed, as are fluctuated greatly
in the experiment. The retained ions were used in downstream statistical analysis. STATISTICAL ANALYSIS All statistical analyses were performed using the Wilcoxon rank-sum test in R (version
4.0.0). DATA RECORDS The rRNA-depleted RNA-seq data generated in this study are available in the NCBI SRA database under accession number SRP39858539. Raw lipidomics data files from each
ionization mode (positive and negative) were deposited at the MetaboLights database with the accession number MTBLS594340. Each MetaboLights entry contains protocols about sample collection,
extraction, chromatography, mass spectrometry, metabolite identification, and data transformation. Other data that support the findings in ‘Technical Validation’ have been deposited in the
Figshare41,42,43,44 repository: (1) Detailed information on single-copy orthologous PCGs across 15 vertebrate species41; (2) PCA plot of PC1 versus PC3 and PC2 versus PC3 based on the
expression levels of single-copy orthologous PCGs among 15 vertebrate species42; (3) Comprehensive information of all identified lipid metabolites43; (4) Identified lipid metabolites in the
negative and positive ion modes44. TECHNICAL VALIDATION TRANSCRIPTOMIC DATA The quality of the RNA-seq data for each sample is shown in Fig. 2. Briefly, a total of ~1.36 terabases (Tb) of
raw data were obtained, which is approximately 12.69 Gb per sample on average (Fig. 2a and Supplementary Table 1). The base quality of the sequencing reads was confirmed using FastQC, and
consisted of a high-quality score Q30 (base error <0.1%) (median Q30 = 92.81%) (Fig. 2a and Supplementary Table 1). After filtering reads for quality and length, we retained a total of
~1.32 Tb of high-quality data, with an average of 12.30 Gb per sample (Fig. 2a and Supplementary Table 1). This allowed us to map an average of 91.64% high-quality reads to the respective
genomes of the different species (Fig. 2a and Supplementary Table 1), indicating the reliability of the sequencing data. Using a detection threshold of >0.5 TPM across all replicates in
at least one AT to identify transcribed PCGs for each species, we observed comparable amounts of transcribed PCGs between mammal (~12,324 per species) and bird species (~11,973 per species)
(Table 1). However, remarkably few transcribed genes were detected in the ATs of turtle (4037 PCGs), implying the AT transcriptomes of mammals and birds vary significantly from reptiles. To
assess the reproducibility of the different biological replicates, we calculated pairwise Spearman’s _r_ of PCGs expression profiles between the samples of each species. In general, gene
expression was highly correlated between biological replicates (median Spearman’s _r_ = 0.96), and similarities significantly reduced between the ATs obtained from different anatomical
locations within each species (_P_ = 9.76 × 10−16, Wilcoxon rank-sum test) (Fig. 2b). Principal component analysis and hierarchical clustering of the expression levels of 3878 single-copy
orthologous PCGs (detailed information is list in file ‘Detailed information on single-copy orthologous PCGs across 15 vertebrate species’ on Figshare41) among 15 vertebrates revealed that
samples primarily clustered according to the species with the highest number of biological replicates (Fig. 2c,d and figure ‘PCA plot of PC1 versus PC3 and PC2 versus PC3 based on the
expression levels of single-copy orthologous PCGs among 15 vertebrate species’ on Figshare42). These results highlight the consistency among biological replicates and the robustness of
experimental design. Finally, to validate evolutionary signatures, we conducted neighbor-joining (NJ) tree analysis based on the expression levels of 3825 transcribed single-copy orthologous
PCGs (Fig. 2e), which was highly consistent with the phylogenetic tree downloaded from the Timetree database (http://www.timetree.org/) (Fig. 1). Moreover, we observed that transcriptional
conservation decreases with evolutionary distance between the species (Fig. 2f), again confirming the reliability of the evolutionary signature uncovered in our dataset. LIPIDOMIC DATA
Lipids concentrations in 131 ATs samples obtained from five vertebrate species were analyzed using LC-MS/MS in positive and negative ion modes. After normalizing the bias within and between
batches using the QC-RSC method38 (Fig. 3a), a total of 2652 negative ions and 4530 positive ions were detected in >50% of the QC samples (102 QC samples in negative ion mode and 96 QC
samples in positive ion mode) and >20% of the experimental samples in at least one AT. To ensure the reliability of the acquired lipidomics data, two quality control steps were performed
based on the intensity of the detected ions. First, we assessed the clustering of QC samples using principal component analysis (PCA). In both ion modes, we observed that the QC samples
clustered distinctively (Fig. 3b), suggesting absence of significant non-biological induced variation in the experiment. Second, we measured the relative standard deviation (RSD) of detected
ions in all QC samples (Fig. 3c). We found that most of the negative (67.42%, 1788 of 2652) and positive ions (81.15%, 3676 of 4530) had an RSD < 30% among the QC samples, indicating ion
intensity changed little between QC samples. Overall, these findings confirmed the reproducibility and robustness of the generated lipidomics data. To further characterize lipid patterns
and evaluate the presence of abnormal samples, we performed _t_-distributed stochastic neighbor embedding (_t_-SNE) analysis (Fig. 4a) based on the intensity of ions with RSD <30% among
the QC samples, and observed a clear separation between distinct species. We next calculated the distance between samples using _t_-SNE to quantify sample variance, which also revealed that
biological replicates tend to cluster close to each other. Moreover, we found that smaller distances between ATs within a species and higher between species (Fig. 4b), highlighting large
variation between different species. Finally, we annotated the ions and identified a total of 868 and 293 lipid metabolites in the positive and negative ion modes, respectively (details in
file ‘Comprehensive information of all identified lipid metabolites’ on Figshare43). These lipid-related metabolites were part of seven lipid categories, including Fatty Acyls (FA),
Glycerolipids (GL), Glycerophospholipids (GP), Prenol lipids (PR), Saccharolipids (SL), Sphingolipids (SP), Sterol Lipids (ST) (details in ‘Identified lipid metabolites in the negative and
positive ion modes’ on Figshare44). USAGE NOTES The AT transcriptomes across the vertebrate species used in this study were profiled using an rRNA-depleted protocol, which enables the
capture of different RNA species and is more efficient in quantifying linear non-poly(A) transcripts and circular RNAs45,46. This will enable the investigation of dynamic PCGs expression
across species, but also comparative analysis of regulatory non-protein coding transcripts (such as long non-coding RNAs) between ATs in vertebrates. Notably, both the AT transcriptome and
lipidome varied more between than within species, which highlights the functional differences that occurred over the course of evolution. This large-scale dataset encompasses a variety of
species with remarkably divergent phylogenetic relationships and ATs with highly diverse anatomical distributions, documenting more than 300 million years of AT evolutionary history. This
valuable resource will make it possible to understand the molecular basis of these functional differences. Theoretically, cross-species comparisons should be conducted using adipose tissues
with similar anatomical locations across species. However, it is not possible to match all adipose tissues between different species. For example, there is no human equivalent of murine
gonadal adipose tissue (the most commonly used visceral adipose tissue in mouse studies). This calls for omics-based comparative analysis to explore how the adipose pads of one species may
correspond to the adipose depots of another species. Indeed, the assessment of human–animal relationships is important for translational studies. We believe that our data will also help to
identify which species should be used in specific settings or as an animal model. For the vast majority of the analyzed species, the AT samples were derived from multiple anatomical
locations, which can be divided into SAT and VAT, allowing for evaluation of transcriptomic and lipidomic similarities and differences among ATs within species, including lesser
characterized non-model organisms. They could also provide an opportunity to investigate the differences between AT locations from an evolutionary point of view. Finally, our multi-omics
data can facilitate more precise investigations of the interaction between mRNAs, non-coding transcripts and metabolites across vertebrate ATs or within species across AT locations. CODE
AVAILABILITY The following software and versions were used for quality control and data processing: (1) RNA-seq data processing: read mapping was performed with the STAR alignment tool
(2.5.3a)31; quantification of RNA-seq data was performed using Kallisto (0.44.0)32; identification of single-copy orthologous genes was performed using OrthoMCL47. (2) Lipidomics data
processing: the raw data were processed using the Progenesis QI software (version 2.2, Nonlinear dynamics) for peak detection and alignment; The metaX software34 was further used to process
peak intensity data. Associated codes have been submitted in GitHub (https://github.com/JiamanZhang/Lab_cross_15_vertebrates_adiposes_papre_code). REFERENCES * Rosen, E. D. & Spiegelman,
B. M. Adipocytes as regulators of energy balance and glucose homeostasis. _Nature._ 444, 847–853 (2006). Article CAS PubMed PubMed Central ADS Google Scholar * Rosen, E. D. &
Spiegelman, B. M. What we talk about when we talk about fat. _Cell._ 156, 20–44 (2014). Article CAS PubMed PubMed Central Google Scholar * Fruhbeck, G. Overview of adipose tissue and
its role in obesity and metabolic disorders. _Methods Mol. Biol._ 456, 1–22 (2008). Article PubMed Google Scholar * Zwick, R. K., Guerrero-Juarez, C. F., Horsley, V. & Plikus, M. V.
Anatomical, physiological, and functional diversity of adipose tissue. _Cell Metab._ 27, 68–83 (2018). Article CAS PubMed PubMed Central Google Scholar * Dodson, M. V. _et al_. Adipose
depots differ in cellularity, adipokines produced, gene expression, and cell systems. _Adipocyte._ 3, 236–241 (2014). Article CAS PubMed PubMed Central Google Scholar * Morigny, P.,
Boucher, J., Arner, P. & Langin, D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. _Nat. Rev. Endocrinol._ 17, 276–295 (2021). Article CAS
PubMed Google Scholar * Tchkonia, T. _et al_. Mechanisms and metabolic implications of regional differences among fat depots. _Cell Metab._ 17, 644–656 (2013). Article CAS PubMed PubMed
Central Google Scholar * Wajchenberg, B. L. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. _Endocr. Rev._ 21, 697–738 (2000). Article CAS PubMed
Google Scholar * Ottaviani, E., Malagoli, D. & Franceschi, C. The evolution of the adipose tissue: a neglected enigma. _Gen. Comp. Endocrinol._ 174, 1–4 (2011). Article CAS PubMed
Google Scholar * Pond, C. M. An evolutionary and functional view of mammalian adipose tissue. _Proc. Nutr. Soc._ 51, 367–377 (1992). Article CAS PubMed Google Scholar * Sun, W. _et al_.
snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. _Nature._ 587, 98–102 (2020). Article CAS PubMed ADS Google Scholar * Crippa, S. _et al_. Comparative
transcriptome profiling of the injured zebrafish and mouse hearts identifies miRNA-dependent repair pathways. _Cardiovasc. Res._ 110, 73–84 (2016). Article CAS PubMed PubMed Central
Google Scholar * Baboota, R. K. _et al_. Microarray based gene expression analysis of murine brown and subcutaneous adipose tissue: significance with human. _PLoS ONE._ 10, e0127701 (2015).
Article PubMed PubMed Central Google Scholar * Zuriaga, M. A., Fuster, J. J., Gokce, N. & Walsh, K. Humans and mice display opposing patterns of “browning” gene expression in
visceral and subcutaneous white adipose tissue depots. _Front. Cardiovasc. Med._ 4, 27 (2017). Article PubMed PubMed Central Google Scholar * Emont, M. P. _et al_. A single-cell atlas of
human and mouse white adipose tissue. _Nature._ 603, 926–933 (2022). Article CAS PubMed PubMed Central ADS Google Scholar * _NCBI Sequence Read Archive_
https://www.ncbi.nlm.nih.gov/sra/SRP292518 (2022). * _GSA for human_ https://ngdc.cncb.ac.cn/gsa-human/browse/HRA002514 (2022). * Jin, L. _et al_. A pig bodymap transcriptome reveals diverse
tissue physiologies and evolutionary dynamics of transcription. _Nat. Commun._ 12, 3715 (2021). Article CAS PubMed PubMed Central ADS Google Scholar * _NCBI Sequence Read Archive_
https://www.ncbi.nlm.nih.gov/sra/SRP266207 (2021). * _NCBI Sequence Read Archive_ https://www.ncbi.nlm.nih.gov/sra/SRP321725 (2022). * _NCBI Gene Expression Omnibus_,
https://identifiers.org/geo/GSE199968 (2023). * Leiria, L. O. & Tseng, Y. H. Lipidomics of brown and white adipose tissue: implications for energy metabolism. _BBA-Mol_. _Cell Biol. L._
1865, 158788 (2020). CAS Google Scholar * Yore, M. M. _et al_. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. _Cell._ 159, 318–332
(2014). Article CAS PubMed PubMed Central Google Scholar * Marcher, A. B. _et al_. RNA-seq and mass-spectrometry-based lipidomics reveal extensive changes of glycerolipid pathways in
brown adipose tissue in response to cold. _Cell Rep._ 13, 2000–2013 (2015). Article CAS PubMed Google Scholar * Grzybek, M. _et al_. Comprehensive and quantitative analysis of white and
brown adipose tissue by shotgun lipidomics. _Mol. Metab._ 22, 12–20 (2019). Article CAS PubMed PubMed Central Google Scholar * May, F. J. _et al_. Lipidomic adaptations in white and
brown adipose tissue in response to exercise demonstrate molecular species-specific remodeling. _Cell Rep._ 18, 1558–1572 (2017). Article CAS PubMed PubMed Central Google Scholar *
Chondronikola, M. _et al_. Brown adipose tissue activation is linked to distinct systemic effects on lipid metabolism in humans. _Cell Metab._ 23, 1200–1206 (2016). Article CAS PubMed
PubMed Central Google Scholar * Wang, W. C. _et al_. Lipidomic profiling of high-fat diet-induced obesity in mice: importance of cytochrome P450-derived fatty acid epoxides. _Obesity_ 25,
132–140 (2017). Article PubMed Google Scholar * Hou, B. _et al_. Targeted lipidomics and transcriptomics profiling reveal the heterogeneity of visceral and subcutaneous white adipose
tissue. _Life Sci._ 245, 117352 (2020). Article CAS PubMed PubMed Central Google Scholar * Li, Y. _et al_. Pacific Biosciences assembly with Hi-C mapping generates an improved,
chromosome-level goose genome. _GigaScience._ 9, giaa114 (2020). Article PubMed PubMed Central Google Scholar * Dobin, A. _et al_. STAR: ultrafast universal RNA-seq aligner.
_Bioinformatics._ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. _Nat.
Biotechnol._ 34, 525–527 (2016). Article CAS PubMed Google Scholar * Huang, C. _et al_. Analysis of lipidomics profile of Carya cathayensis nuts and lipid dynamic changes during
embryonic development. _Food Chem._ 370, 130975 (2022). Article CAS PubMed Google Scholar * Wen, B., Mei, Z., Zeng, C. & Liu, S. metaX: a flexible and comprehensive software for
processing metabolomics data. _BMC Bioinformatics._ 18, 1–14 (2017). Article Google Scholar * Troyanskaya, O. _et al_. Missing value estimation methods for DNA microarrays.
_Bioinformatics._ 17, 520–525 (2001). Article CAS PubMed Google Scholar * Gromski, P. S. _et al_. Influence of missing values substitutes on multivariate analysis of metabolomics data.
_Metabolites._ 4, 433–452 (2014). Article PubMed PubMed Central Google Scholar * Dieterle, F., Ross, A., Schlotterbeck, G. & Senn, H. Probabilistic quotient normalization as robust
method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. _Anal. Chem._ 78, 4281–4290 (2006). Article CAS PubMed Google Scholar * Kirwan, J. A.,
Broadhurst, D. I., Davidson, R. L. & Viant, M. R. Characterising and correcting batch variation in an automated direct infusion mass spectrometry (DIMS) metabolomics workflow. _Anal.
Bioanal. Chem._ 405, 5147–5157 (2013). Article CAS PubMed Google Scholar * _NCBI Sequence Read Archive_ https://identifiers.org/ncbi/insdc.sra:SRP398585 (2022). * Liu, P. _et al_.
Transcriptomic and lipidomic profiling of subcutaneous and visceral adipose tissues in 15 vertebrates. _MetaboLights_ https://www.ebi.ac.uk/metabolights/MTBLS5943 (2022). * Liu, P. _et al_.
Detailed information on single-copy orthologous PCGs across 15 vertebrate species. _figshare._ https://doi.org/10.6084/m9.figshare.22656418.v2 (2023). * Liu, P. _et al_. PCA plot of PC1
versus PC3 and PC2 versus PC3 based on the expression levels of single-copy orthologous PCGs among 15 vertebrate species. _figshare._ https://doi.org/10.6084/m9.figshare.22656454.v2 (2023).
* Liu, P. _et al_. Comprehensive information of all identified lipid metabolites. _figshare._ https://doi.org/10.6084/m9.figshare.22643428.v2 (2023). * Liu, P. _et al_. Identified lipid
metabolites in the negative and positive ion modes. _figshare._ https://doi.org/10.6084/m9.figshare.22657012.v3 (2023). * Katayama, S. _et al_. Antisense transcription in the mammalian
transcriptome. _Science._ 309, 1564–1566 (2005). Article PubMed ADS Google Scholar * Chen, L. _et al_. Paired rRNA-depleted and polyA-selected RNA sequencing data and supporting
multi-omics data from human T cells. _Sci. Data._ 7, 1–7 (2020). Article ADS Google Scholar * Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups
for eukaryotic genomes. _Genome Res._ 13, 2178–2189 (2003). Article CAS PubMed PubMed Central Google Scholar * Saeed, A. I. _et al_. TM4: a free, open-source system for microarray data
management and analysis. _Biotechniques._ 34, 374–378 (2003). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank the High-Performance Computing Platform of
Sichuan Agricultural University for providing computing resources and support that have contributed to this research. This work was supported by the National Key R & D Program of China
(2020YFA0509500 and 2021YFA0805903), the Sichuan Science and Technology Program (2021ZDZX0008 and 2021YFYZ0009), the National Natural Science Foundation of China (32225046, 32102507,
32102512 and U22A20507). AUTHOR INFORMATION Author notes * These authors contributed equally: Pengliang Liu, Diyan Li. AUTHORS AND AFFILIATIONS * School of Pharmacy, Chengdu University,
Chengdu, 610106, China Pengliang Liu & Diyan Li * Livestock and Poultry Multi-omics Key Laboratory of Ministry of Agriculture and Rural Affairs, College of Animal Science and Technology,
Sichuan Agricultural University, Chengdu, 611130, China Pengliang Liu, Jiaman Zhang, Rui Liu & Mingzhou Li * Animal Breeding and Genetics Key Laboratory of Sichuan Province, Institute
of Animal Genetics and Breeding, Sichuan Agricultural University, Chengdu, 611130, China Pengliang Liu, Jiaman Zhang, Rui Liu & Mingzhou Li * Chengdu Research Base of Giant Panda
Breeding, Chengdu, 611081, China Mengnan He & Yan Li Authors * Pengliang Liu View author publications You can also search for this author inPubMed Google Scholar * Diyan Li View author
publications You can also search for this author inPubMed Google Scholar * Jiaman Zhang View author publications You can also search for this author inPubMed Google Scholar * Mengnan He View
author publications You can also search for this author inPubMed Google Scholar * Yan Li View author publications You can also search for this author inPubMed Google Scholar * Rui Liu View
author publications You can also search for this author inPubMed Google Scholar * Mingzhou Li View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS M.L. and D.L. conceived and designed the study. M.H., R.L. and P.L. performed animal work and sample preparation. M.H. and P.L. constructed the RNA-seq libraries. R.L. and P.L.
performed LC-MS/MS data acquisition. J.Z. and Y.L. performed the data processing and analysis. J.Z. and P.L. performed the statistical analysis. J.Z. and P.L. visualized data. P.L. and D.L.
wrote the manuscript. M.L. revised the paper. CORRESPONDING AUTHORS Correspondence to Diyan Li or Mingzhou Li. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY TABLE 1. SUMMARY OF ALL RNA-SEQ DATA ACROSS 15 VERTEBRATE SPECIES. SUPPLEMENTARY TABLE 2. SUMMARY OF THE SAMPLES USED IN THE LIPIDOMICS STUDY. RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liu, P., Li, D.,
Zhang, J. _et al._ Transcriptomic and lipidomic profiling of subcutaneous and visceral adipose tissues in 15 vertebrates. _Sci Data_ 10, 453 (2023).
https://doi.org/10.1038/s41597-023-02360-3 Download citation * Received: 10 October 2022 * Accepted: 03 July 2023 * Published: 12 July 2023 * DOI: https://doi.org/10.1038/s41597-023-02360-3
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative