- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:
ABSTRACT Multidrug resistance-associated protein 2 (MRP2/ABCC2) is a polyspecific efflux transporter of organic anions expressed in hepatocyte canalicular membranes. MRP2 dysfunction, in
Dubin-Johnson syndrome or by off-target inhibition, for example by the uricosuric drug probenecid, elevates circulating bilirubin glucuronide and is a cause of jaundice. Here, we determine
the cryo-EM structure of rat Mrp2 (rMrp2) in an autoinhibited state and in complex with probenecid. The autoinhibited state exhibits an unusual conformation for this class of transporter in
which the regulatory domain is folded within the transmembrane domain cavity. In vitro phosphorylation, mass spectrometry and transport assays show that phosphorylation of the regulatory
domain relieves this autoinhibition and enhances rMrp2 transport activity. The in vitro data is confirmed in human hepatocyte-like cells, in which inhibition of endogenous kinases also
reduces human MRP2 transport activity. The drug-bound state reveals two probenecid binding sites that suggest a dynamic interplay with autoinhibition. Mapping of the Dubin-Johnson mutations
onto the rodent structure indicates that many may interfere with the transition between conformational states. SIMILAR CONTENT BEING VIEWED BY OTHERS STRUCTURAL BASIS FOR THE TRANSPORT AND
REGULATION MECHANISM OF THE MULTIDRUG RESISTANCE-ASSOCIATED PROTEIN 2 Article Open access 08 January 2025 EXTRACELLULAR MUTATION INDUCES AN ALLOSTERIC EFFECT ACROSS THE MEMBRANE AND HAMPERS
THE ACTIVITY OF MRP1 (ABCC1) Article Open access 08 June 2021 STRUCTURAL INSIGHTS INTO HUMAN ORGANIC CATION TRANSPORTER 1 TRANSPORT AND INHIBITION Article Open access 15 March 2024
INTRODUCTION MRP2 (multidrug resistance-associated protein, 2) is a primary active transporter of the ATP-binding cassette (ABC) class1. There are nine C-subfamily members in humans with
distinct physiological roles ranging from a chloride channel (the cystic fibrosis transmembrane regulator (CFTR); ABCB7) and a regulator of potassium channels (the sulphonyl urea receptors 1
and 2; ABCC8 and 9) to exporters of organic anions (MRP1-5). MRP2 is localised to the apical membranes of polarised cells such as hepatocytes, enterocytes, pneumocytes and proximal tubule
cells of the kidney. Loss of MRP2 function manisfests primarily as a liver disease caused by the failure to export bilirubin glucuronide, the hepatic product of heme breakdown. Failure to
eliminate the compound across the canalicular membrane into the bile ultimately results in jaundice, the primary feature of Dubin–Johnson syndrome, after the conjugated bilirubin is exported
back into the circulation by the basolateral MRP32. Over 40 mutations have been identified in the _ABCC2_ gene, some of which are neutral, others are non-sense or missense mutations causing
the absence of the MRP2 protein from the hepatocyte apical membrane or MRP2 with compromised transport activity, respectively3,4. Other transport substrates of MRP2 have been identified in
vitro and include glutathione- sulfate-, and glucuronide- conjugated metabolites of Phase II metabolism. These include conjugated leukotriene C4 (conjugated LTC4) and therapeutic anticancer
drugs such as paclitaxel and cisplatin5,6,7,8,9,10,11. The physiological relevance of drug transport remains unclear but animal studies have shown that engineered expression of MRP2 in
implanted cancer cells confers resistance to cisplatin12 and in a small clinical study MRP2 expression levels in hepatocellular cancer patients correlated with a reduction in
cisplatin-induced tumour necrosis suggesting that MRP2 is a potential determinant of multidrug resistance13. The activity of several ABCC transporters are known to be mediated by protein
kinases. For example, phosphorylation of MRP1 by the protein kinase CK2 alpha (CK2α) leads to increased efflux of doxorubicin14, while CFTR is activated by protein kinases C and A to allow
ATP-dependent channel opening15,16. In addition to phosphorylation, transport activity can also be modulated by drug-drug interactions. For MRP2 the uricosuric agent probenecid and the
diuretic furosemide have been shown to inhibit the efflux of N-ethylmaleimide glutathione in vitro17. Jaundice is also a reported off-target side effect of probenecid18. In this work, we
screen for mammalian homologues of human MRP2 and we identify the _Rattus norvegicus_ Mrp2 (rMrp2) as suitable for structural studies (80% sequence identity to its human ortholog). We
determine the cryo-EM structure of rMrp2 in a nucleotide-free state (3.21 Å resolution) and a drug-bound state (3.45 Å resolution) with the uricosuric agent probenecid. The nucleotide-free
state is in an autoinhibited state. Functional data coupled to mass spectrometry-based proteomics identify phosphorylation sites that likely modulate the activation of the protein and
transport data with rMrp2 reconstituted in liposomes show a strong correlation between the phosphorylation state of the transporters and transport activity. This observation is corroborated
in a cellular system using human hepatocytes derived from induced-pluripotent stem cells (iPSCs) which shows a reduction in MRP2 transport activity following inhibition of protein kinase
activity. The probenecid bound structure reveals two drug binding sites within the rMrp2 cavity. The mutations that cause Dubin–Johnson syndrome are mapped onto the rMrp2 structure, which
provides insights on how they contribute to MRP2 dysfunction by interfering with conformational changes along the transport cycle rather than substrate binding. Based on this work, we
propose a mechanism for the modulation of the MRP2 activity by phosphorylation and drugs. RESULTS CRYO-EM STRUCTURE OF NUCLEOTIDE-FREE RMRP2 We expressed rMrp2 in _Saccharomyces cerevisiae_
as part of our screen to identify homologues suitable for structural studies19. Extraction of rMrp2 with Lauryl Maltose Neopentyl Glycol (LMNG) and cholesteryl hemisuccinate (CHS) and
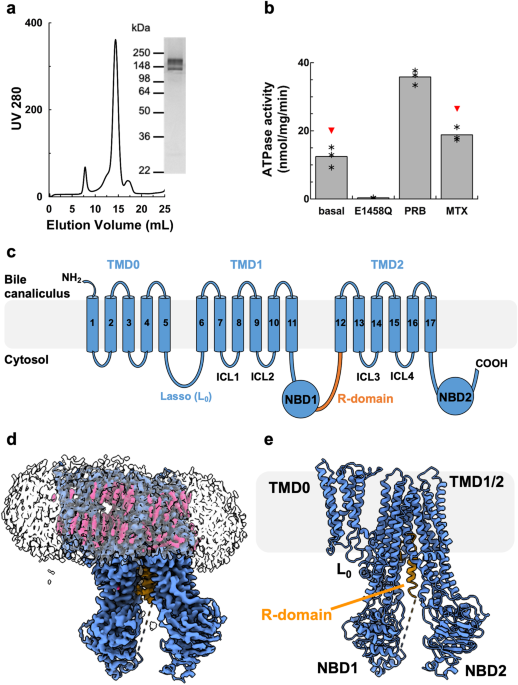
further purification into glyco-diosgenin (GDN) resulted in highly pure and monodisperse sample suitable for functional and structural studies (Fig. 1). The recombinant rMrp2 was
reconstituted in destabilised liposomes consisting of bovine liver lipid extract, and it displayed a basal ATPase activity of 12.43 ± 3 nmol/mg/min that could be stimulated by the clinical
drugs probenecid and methotrexate by 2.9 and 1.5 fold, respectively17 (Fig. 1). Single-particle cryo-EM analysis of the nucleotide-free rMRP2 resulted in a three-dimensional reconstruction
at an overall resolution of 3.21 Å (Fourier shell correlation (FSC) = 0.143 criterion; Supplementary Fig. 1 and Supplementary Table 1). The maps showed density with good connectivity and the
presence of side chains that could be easily interpreted for model building. The rMrp2 (AF-Q63120-F1) AlphaFold2 model was used as a starting model. We have built an atomic model for rMrp2
that consists of a small N-terminal transmembrane domain (TMD0) that is linked to two transmembrane domains (TMD), TMD1 and TMD2, by the lasso linker (L0). TMD0 is composed of a bundle of 5
transmembrane helices (TMs); the interface of TMD0 and TMD1 is occupied by several CHS molecules (not modelled as only part of the sterol moiety was resolved) (Fig. 1). The L0 linker has
been shown to facilitate correct folding and trafficking of ABCC transporters20,21. Each TMD consists of 6 TM helices, enclosing the pocket for substrates and drugs binding, and they are
linked to two nucleotide binding domains (NBDs), NBD1 and NBD2, where ATP binding and hydrolysis provides the free energy required for the transport cycle completion. NBD1 and NBD2 form an
interface with the TMD1 and TMD2 via the intracellular loop 4 (ICL4) and 2 (ICL2), respectively; ICL2 and ICL4 are known as coupling helices and their role is to transmit conformational
changes to the TMDs, associated with either NBD dimerisation or disengagement upon ATP binding or hydrolysis, respectively22. In the absence of nucleotides, rMrp2 adopts an inward-facing
conformation with the TMDs open to the cytosol where substrates and drugs can access it. An unusual feature of the rMrp2 cryo-EM maps was the presence of helical-like density within the TMD1
and TMD2 interface that corresponds to the regulatory domain (R-domain) (Fig. 2); MRP2 and other members of the ABCC family contain an R-domain that regulates their transport activity upon
phosphorylation by specific kinases (CKII, PKC, PKA and PLK kinases)23. The R-domain is predicted to be partly helical from the AlphaFold2 model and it links NBD1 to the elbow helix (short
helix at the amine-terminal of TM12 and it sits perpendicular to the TMD2), but in the predicted model it was placed at the interface of the NBDs and outside the TMD whereas in our structure
it sits deep inside the TMD. The R-domain consists of residues Gly863-Thr958. Although, continuous density from NBD1 to the helical density can be observed, only the helical part, residues
Ala895-Lys924, was modelled as the rest of the density is weak and without defined side chains; in addition, there is no density that links the C-terminus of the R-domain to the elbow helix.
The R-domain is buried within the TMDs at the interface of TMs 8, 9, 11 and 17 and it is stabilised by several hydrogen bonds and Van der Waals interactions (Fig. 2). The overall
architecture of rMrp2 resembles that of another member of the ABCC family, the bovine Mrp1 (bMrp1)24. MRP1 is the closest homologue of MRP2 (49% identity between the human sequences) but it
is localised in the basolateral membrane of polarised cells in most tissues with relatively low levels in liver. Although MRP1 and MRP2 have some common substrates, there are significant
differences on clinical drug specificity25. Despite their overall fold being very similar, rMrp2 and bMrp1 can be superimposed with a root-mean-square deviation (rmsd) of 5.6 Å over 1400 Cα
atoms as a result of a 26° tilt of the TMD0 away from TMD1 (Supplementary Fig. 2); excluding the TMD0 from the alignment, the two structures can be superimposed with an rmsd of 3 Å. The main
conformational difference is in the NBDs, that adopt a more closed conformation in rMrp2 due to a 2.5 Å displacement of the NBD1 in tandem with ICL4 towards NBD2 (Supplementary Fig. 2). Two
other members of the ABCC family that have their structures resolved are the human cystic fibrosis transmembrane conductance regulator (CFTR)26 and the _S. cerevisiae_ Ycf1
transporter27,28, but these are functionally distinct from rMrp2 functioning as a chloride channel and cadmium metal transporter, respectively. The most striking difference between rMrp2,
CFTR and Ycf1 is the conformation of the R-domain; the R-domain in bMrp1 was not resolved. In CFTR, the dephosphorylated R-domain adopts a helical structure similar to rMrp2 (Fig. 2); in the
CFTR structure, a 19 residue helix, presumed to be from the R domain, is resolved and it is localised in the cytosolic face of the TMDs making contact with the intracellular regions of TMs
14 and 15. The CFTR is in an inactive-like conformation as the R-domain sits between NBD1 and NBD2 and prevents them from forming an interface26. In phosphorylated Ycf1, 30 residues of the
R-domain were resolved, it adopts an extended conformation outside the interface of TMD1/2 where it is stabilised by residues on the surface of NBD127,28. In the phosphorylated CFTR the
R-domain retains its helical structure and it is stabilised by interactions with residues from TMs 14, 15 and 1729 (Fig. 2). As the R-domain of rMrp2 is deep inside the TMD1/2 interface, it
points to an inactive conformation that would prevent bilirubin glucuronide or drug binding and the formation of an NBD:NBD interface. This is similar to the situation with CFTR where the
weak density that links NBD1 and the helical portion of the R-domain is found close to NBD1 that would prevent it from interacting with NBD2 upon ATP binding, due to steric clashes.
REGULATION BY PHOSPHORYLATION Considering the conformation of the R-domain in rMrp2, we investigated the phosphorylation state of the purified protein and probed how phosphorylation
regulates its activity. The analysis of the purified rMrp2 by SDS-PAGE phosphostaining showed that the recombinant protein is phosphorylated after purification (Fig. 3). Mass
spectrometry-based proteomics analysis of the purified rMrp2 provided more quantitative data; it showed that rMrp2 is phosphorylated at several predicted sites (Fig. 3, Supplementary Fig. 3
and Source Data file), including residues of the R-domain, Thr869, Ser874 and Thr886, with the exception of Ser922 (only a very weak ambiguous signal could be detected) and Ser926 (Fig. 3);
the partially phosphorylated protein will be referred as rMrp2R-*. The R-domain contains several phosphorylation motifs for the CKII, PKC and PLK kinases30. Although, similar kinases are
present in _S. cerevisiae_, it is very likely that subtle changes in the sequence or conformation of rMrp2 may prevent phosphorylation of all the predicted sites. In our cryo-EM maps, we did
not resolve any phospho modified residues. We manipulated the phosphorylation state of rMrp2 in vitro, to determine the impact of phosphorylation on its activity. Firstly, we fully
dephosphorylated Mrp2R-* with λ-protein phosphatase (referred as rMrp2R-de); dephosphorylation did not alter the ATPase activity of rMrp2 in destabilised liposomes (Fig. 3). Since _S.
cerevisiae_ is not capable of fully phosphorylating the R-domain of rMrp2, we devised a protocol for the in vitro phosphorylation of rMrp2R-* during purification. We incubated rMrp2R-* with
a cell extract from human embryonic kidney 293 (HEK293) cells that contains the endogenous kinases, in the presence of phosphatase inhibitors to prevent dephosphorylation by endogenous
phosphatases. The rMrp2 was further purified to remove the cell extract proteins. Phosphostaining showed a 48% enhancement of rMrp2 phosphorylation compared to rMrp2R-* using this in vitro
protocol (Fig. 3). Quantitative analysis by mass spectrometry confirmed that in the presence of the extract, rMrp2 is fully phosphorylated including Ser922 and Ser926 in the R-domain in
addition to other sites throughout the transporter (referred as rMrp2R-pho) (Fig. 3). The basal ATPase activity of rMrp2R-pho showed 70% activity enhancement relative to the rMrp2R-* and
rMrp2R-de and inclusion of probenecid in the assays did not stimulate the ATPase activity any further suggesting a critical role for Ser922 and Ser926 on modulating the activity of rMrp2.
Since the different levels of rMrp2 phosphorylation impacted the ATPase activity, we also investigated their influence in its transport activity. rMrp2 was reconstituted in proteolisomes and
we measured the rMrp2-dependent uptake of the fluorescent substrate 5(6)-Carboxy-2′,7′-dichlorofluorescein (CDF) inside the liposomes. The Mrp2R-* and rMrp2R-de transported the CDF with a
rate of 0.5 nmol/mig/30 min whereas the rMrp2R-pho displayed a significant increase in its transport rate of 3 nmol/mig/30 min. The increased basal ATPase activity and uptake of the CDF by
the fully phosphorylated rMrp2 suggested that full phosphorylation of the R-domain relieves its inhibitory effect and prevents it from occupying the TMD1/2 interface; a similar effect was
observed for the fully phosphorylated CFTR whose channel opening and ATPase activity were enhanced upon phosphorylation26. To further assess the effect of phosphorylation on the activity of
MRP2 in a cellular context, we performed transport assays in polarised hepatocyte-like cells derived from human induced pluripotent stem cells (iPSCs) (induced hepatocytes (iHEPs))
expressing human MRP2 in the absence and presence of the broad-spectrum kinase inhibitor staurosporine, which should prevent human MRP2 phosphorylation by the endogenous kinases. The iHEPs
are polarised, make and secrete bile into canaliculi formed between adjacent cells, thus mimicking the native cellular context of human MRP2. Human MRP2 can transport the CDF into the bile
canaliculi that is reduced by nearly 50% in the presence of staurosporine (Fig. 4) further inferring the role of phosphorylation in modulating the activity of MRP2 (treatment of the iHEPs
with staurosporine does not alter the localisation or expression levels of human MRP2 (Supplementary Fig. 4). Our transport data in proteoliposomes and iHEPs provide strong evidence how
phosphorylation of the R-domain impacts substrate transport. DRUG-BOUND RMRP2 STRUCTURE To gain an understanding of the molecular basis of drug recognition and polyspecificity by Mrp2 and
its modulation by drugs, we determined its cryo-EM structure in the presence of probenecid, to 3.45 Å resolution (Fig. 5 and Supplementary Fig. 5). Probenecid is reported to act
idiosynchratically on MRP2, inhibiting the transport of some substrates and stimulating the transport of others5,17. In our assays, probenecid stimulated the basal ATPase activity of rMrp2
but also acted as an inhibitor for the transport of CDF into liposomes that is consistent with this previously reported modulating activity (Figs. 1b and 3d). The cryo-EM map revealed clear
density for two probenecid molecules and one CHS (Fig. 5). Several biochemical studies had proposed that MRP2 (and MRP1) contain two drug binding sites31,32, but they had not been identified
within the TMDs. Strikingly, the two probenecid molecules and the CHS have displaced the R-domain out of the TMD and occupy a similar space as the R-domain in our apo structure (the same
rMrp2 preparation was used for both structures) (Figs. 5 and 6); no density for the R-domain can be observed in our maps suggesting a high degree of flexibility. Displacement of the R-domain
from the TMD by probenecid results in a more closed overall conformation at the cytosolic half-end of the TMD and NBDs by 8 Å, respectively, whereas the otherhalf of the TMD towards the
membrane, where the drug density is found, displays minimal movement (Fig. 6). The TMD region defined by TMs 7, 8, 16 and 17, form the two probenecid binding sites (Fig. 5). The probenecid
in drug-binding site 1 is coordinated by a hydrogen bond between Gln443 and its carboxylate, a hydrogen bond between Trp1250 and the sulfonamide, and a π-stacking interaction between Phe378
and its benzene ring. The probenecid in site 2 is coordinated by a hydrogen bond between its carboxylate and Arg1201 and a π-stacking interaction between Phe587 and its benzene ring (Fig.
5). The CHS molecule is coordinated mostly by van der Waals interactions and a hydrogen bond between its carboxylate and Gln1030. In the bMrp1 structure in complex with conjugated
leukotriene C4 (LTC4)24, the glutathione (GSH) moiety is coordinated by positively charged residues in the P-pocket and its lipid tail by van der Waals interactions within the H-pocket. In
comparison with our rMrp2 structure, the probenecid binding site 1 overlaps with the P-pocket of bMrp1 whereas the drug binding site 2 is distal to both the drug binding site 1 and LTC4
binding site (Fig. 6); the CHS binding site overlaps with the H-pocket of bMrp1. The drug-bound rMrp2 has a very similar overall conformation to the substrate-bound bMrp1, and the two
structures can be superimposed with an rmsd of 2.8 Å (excluding the TMD0), suggesting that the TMD is not required to undergo significant conformational changes to bind substrates and
drugs;selectivity is probably due to small differences in the amino acids ligning the substrate and drug binding sites. DUBIN–JOHNSON SYNDROME Understanding the pathophysiology of
Dubin–Johnson syndrome can shed light on the mechanism of action of MRP2. Several mutations (nonsense, missense, deletion, splice site) have been identified that result in either
dysfunctional MRP2 or absence of MRP2 expression in the apical membrane3,4,6,33. We mapped the mutations that are associated with Dubin–Johnson syndrome onto the rMrp2 structure (Fig. 7) and
interestingly none are found within the likely substrate binding site of the TMD cavity or R-domain suggesting that the observed dysfunction is not due to loss of bilirubin glucuronide
recognition or regulation by phosphorylation. Most of the mutations associated with Dubin–Johnson syndrome are located in the intracellular loops of the TMDs and at the interface of the ICL4
and NBD1, and ICL2 and NBD2, respectively, suggesting uncoupling of the conformational changes associated with signal transduction between the transport substrate binding site(s) and the
ATP catalytyic cycle (Fig. 7); the interface between the two coupling helices and NBDs is critical to transmit the conformational changes associated with either formation of the NBD:NBD
interface upon ATP binding to transition to an outward open conformation or disengagement of the NBDs to revert into an inward open conformation upon ATP hydrolysis. The most common
mutations are R1150H and I1173F in MRP2 that are frequent in the Iranian-Jewish and Moroccan-Jewish populations that comprise the largest groups of Dubin–Johnson syndrome patients34. The
effect of I1173F is due to low membrane insertion rather than reduced activity. R1150H is found at TM15 (which is likely distant from the transport substrate binding site if extrapolation
from the conjugated bMrp1 LTC4 binding pocket reflects the likely binding site of bilirubin glucuronide in MRP2), suggesting that it may interfere with conformational changes along the TMD
or prevent the R-domain from disengaging from the inhibited state; in the phosphorylated CFTR structure the R-domain contacts TM15. DISCUSSION Our cryo-EM structure established that the
molecular architecture of rMrp2 is very similar to other members of the ABCC family, bMrp1, CFTR and Ycf1, despite their low sequence identity. A common feature between rMrp2 and the other
transporters is their regulation by phosphorylation of the regulatory or R-domain. The concept of modulating the activity of transporters, both ABC- and secondary active transporters, is
well established but the exact molecular mechanism of this process remains obscure. With regard to ABC transporters, our current study together with other studies provides the molecular
basis to understand this regulatory process. Functional and structural studies have proposed that the R-domain interferes with NBD:NBD interaction thus reducing ATP hydrolysis and that this
negative regulation can be relieved by phosphorylation26,29,27. Transporters with a dephosphorylated R-domain can still exhibit ATPase activity, suggesting that the inhibition by the
R-domain is transient. It has been speculated that in the dephosphorylated state, the R-domain is in an equilibrium between inhibited and active-like states that is shifted towards the
active state upon phosphorylation26; this equilibrium was proposed on the basis that dephosphorylated CFTR retained low, but not absent, probability of channel opening, that was stimulated
upon phosphorylation26. Although the R-domain is observed deep inside the TMD of the rMRP2 structure, our functional data show that rMrp2 retains its ATPase activity adding further support
for the concept of the R-domain equilibrium. Our structure provides insights on why phosphorylation of the R-domain would relieve the inhibitory state via a mechanism of R-domain movement
hindrance. Our mass spectrometry data coupled to in vitro phosphorylation with human kinases and biochemical assays, ATPase and substrate transport, showed that upon full phosphorylation of
the R-domain, rMrp2 displays faster ATP hydrolysis and substrate transport relative to the partially phosphorylated protein. Interestingly, the ATPase activity of the fully phosphorylated
rMrp2 could not be further stimulated in the presence of probenecid. This is in contrast to ABC transporters that do not contain an R-domain, whose ATPase activity is greatly modulated by
substrates35,36. It is very likely that if the R-domain is partially phosphorylated, transport substrates or drugs will have to compete with the R-domain for the TMD therefore appearing to
stimulate its basal ATPase activity. Phosphorylation can only happen after the R-domain has transiently moved out of the TMD or the NBD:NBD interface (as proposed for CFTR). Our mass
spectrometry data point to the key role of Ser922 and Ser926 in modulating the activity of rMrp2; the purified rMrp2 had a nearly absent phospho signal for Ser922 and Ser926 that was
significantly increased upon in vitro treatment; in our structure we have partly resolved the R-domain and phosphorylation of Ser922 would cause steric clashes with Asn432 and Tyr433 if the
R-domain were to adopt a similar conformation to the dephosphorylated state; alternatively, phosphorylation could also stabilise the R-domain outside the TMD or NBD:NBD interface in a
similar manner to the phosphorylated CFTR26,29 and Ycf1 structures27,28. Although Ser926 has not been resolved in our structure, it may also contribute to steric clashes as it would still be
located inside the TMD. Since the R-domain in our cryo-EM structure is partially phosphorylated, Thr869, Ser874 and Thr886, it suggests that phosphorylation of these residues does not
relieve autoinhibition (full dephosphorylation of rMrp2 did not alter the ATP hydrolysis kinetics either), but their role might be to stabilise the R-domain as stated above. Our functional
data support this observation, as upon phosphorylation the rMrp2 displays faster ATP turnover and CDF transport activity compared to the partially phosphorylated and fully dephosphorylated
proteins. The observation of the rMrp2 autoinhibited state further supports the notion of R-domain plasticity within the TMD and at the NBD:NBD interface. We show that inhibtion of cellular
kinases resulted in significantly reduced CDF export in iHEPs. Our data provide a strong correlation between the in vitro rMrp2 and _in cellulo_ transport activity of MRP2, as the transport
activity of the dephosphorylated rMrp2 was reduced by nearly 80% and the transport activity of human MRP2 was reduced by 50%, respectively. Overall, the transport activity of MRP2 is likely
to be strongly regulated by the activity of kinases and phosphatases in the canaliculi. Despite our mechanistic insights, the exact interplay of kinases and phosphatases to regulate the
phosphorylation state of MRP2 remains unclear; i.e. the exact conditions and timeframe under which MRP2 will be activated or deactivated within the cellular context. Biochemical evidence for
two drug binding sites in MRP1 and MRP2 has been reported31,32,37,38. We observed two molecules of probenecid in our drug-bound structure of rMrp2. Extrapolating from the bMrp1 structure
with bound conjugated-LTC4 these probenecid sites likely overlap to some extent with the prospective binding site for bilirubin glucuronide in MRP2. The drug and transport substrate binding
sites of MRP1 and MRP2 show some sequence conservation, but also several differences that could explain their differences in drug selectivity. Probenecid binding site 2 of rMrp2 displays
sequence conservation with MRP1, whereas the probenecid binding site 1 is less conserved, suggesting a possible role in the latter for drug selectivity differecens between MRP1 and MRP2.
Interestingly, probenecid is capable of either displacing the unphosphorylated R-domain or preventing it from re-entering the TMD upon the transient movement of the R-domain. Our functional
data show that probenecid can stimulate the ATPase activity of partially phosphorylated rMrp2 and the structure provides insights on the modulation of the rMrp2 activity by drugs in addition
to phosphorylation. The displacement of the R-domain by probenecid relieves the inhibitory state by allowing the NBD1 and NBD2 to come closer, thus likely facilitating ATP binding and
hydrolysis as shown by our ATPase data. We do not yet have an ATP-bound structure of rMrp2 but would anticipate that following release from the autoinhibited state by phosphorylation of the
R-domain, bilirubin glucuronide would be able to occupy the binding cavity and induce a conformational change to allow NBD1 and NBD2 to form an interface to bind and hydrolyse ATP driving
transport substrate efflux and return to the apo state. Further experiments will be required to test this hypothesis. The observed CHS molecule is very likely recruited from the micelle as
there should not be any free CHS during the later stages of purification. In our cryo-EM maps, we observe several CHS molecules in the interface of TMD0 and TMDs 1 and 2 but also a tightly
bound CHS molecule between the elbow helix and TMs 14, 15 and 17, that is close to the lateral opening of the TMD. There is some evidence of cholesterol modulating the transport activity of
MRP2 but not transporting it in HepG2 cells39. A likely role for the CHS might be to either act as a conjugate molecule of drugs such as probenecid or mask the hydrophobic pocket that is not
occupied by probenecid. It has been suggested that cholesterol might be acting as a ‘fill-in’ molecule for the ABC transporters ABCA1 and ABCB1, when they transport small substrates by
filling the empty space within the binding site40. This concept is relatable to rMrp2 in complex with probenecid as the two molecules do not occupy the hydrophobic P-pocket; in bMrp1 the
lipid tail of LTC4 occupies this site and a cholesterol molecule is not required. Another possibility is that CHS has been recruited by probenecid as part of its modulating activity5,17.
Probenecid is an interesting molecule as it can act as a drug modulator or inhibitor and our structure provides insights on its recognition by MRP2. Considering the drug bound structure,
modulation of the activity of Mrp2 by probenecid might be due to the presence of the two drug binding sites. We propose that if probenecid is acting as an inhibitor, it quite likely occupies
both sites whereas as a modulator, another drug could displace the probenecid from the drug binding site 1 that is less conserved in the ABCC family. Our structural work not only addresses
how drug modulators affect its function, but it also has implications in understanding drug-drug interactions. Finally, we start getting an understanding on how mutations in Dubin–Johnson
syndrome result in the dysfunction of MRP2 by interfering/disrupting the TMD and NBD interface rather than loss of substrate (bilirubin glucuronide and ATP) recognition. Overall, we show a
strong interplay of modulating the activity of rMrp2 by phosphorylation and drugs and start gaining insights on how drug modulators, such as probenecid, could influence drug-drug
interactions. METHODS PROTEIN EXPRESSION AND PURIFICATION The _rAbcc2_ gene was cloned into the p424GAL1 vector with a C-terminal TEV protease site upstream of a GFP-His8 and the construct
was transformed into the _S. cerevisiae_ FGY217 strain. Expression was performed as previously described19; in brief wt rMrp2 or E1458Q rMrp2 expressing strains were inoculated in 10 ml of
-URA media (Yeast synthetic drop-out medium supplement without Ura) containing 2% glucose and incubated overnight in an orbital shaker at 280 RPM and 30 °C. The overnight culture was diluted
in 150 mL of the same media and incubated overnight under the same conditions. The second overnight culture was diluted to an OD600 of 0.12 into 1 L of -URA containing 0.1% glucose in a
2.5-L baffled shaker flask and incubated in the same conditions until the culture reached an OD600 of 0.6. Recombinant protein expression was induced by adding 2% galactose and incubated for
22 h. After expression, the cells were harvested, and the cell pellet was flash frozen in liquid nitrogen (LN2) and stored at −80 °C. The isolated membranes were diluted to 360 mL of
Membrane buffer supplemented with 2% LMNG and 0.4% CHS and incubated under mild agitation for 90 min. The unsolubilised fraction was removed by ultracentrifugation at 190,000 × g for 1 h.
The supernatant was loaded onto a HisTrap HP 5 mL Ni affinity column and washed with 5 column volumes (CV) wash buffer I (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 2 mM DTT, 5%
Glycerol, 0.1% LMNG, 0.02% CHS, 70 mM imidazole), followed by 5 CV of wash buffer II (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 2 mM DTT, 5% Glycerol, 0.02% GDN) and eluted with
elution buffer (50 mM tris/HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 2 mM DTT, 5% Glycerol, 0.02% GDN, 250 mM imidazole). The protein solution was concentrated using a 100 kDa concentrator and
back diluted into the same elution buffer but without imidazole to drop the imidazole concentration belove 70 mM. TEV protease was added in a 1:1 molar ratio and incubated under mild
agitation overnight. The protein was loaded onto a HisTrap HP 5 mL Ni affinity column and the flow trough was collected and the column was washed with 5 CV wash buffer II. All the fractions
containing rMrp2 were pulled together and applied onto a Superose 6 gel filtration column (GE Healthcare) equilibrated with SEC buffer (50 mM Tris/HCl pH 8.0, 150 mM KCl, 2 mM MgCl2, 2 mM
DTT, 0.02% GDN). Peak fractions were analysed by SDS-PAGE, concentrated to 5 mg/ml and were either used immediately to prepare cryo-EM grids or flash frozen in LN2 for assays. PROTEIN
PHOSPHORYLATION AND DEPHOSPHORYLATION Purified rMrp2R-* was either treated with λ-protein phosphatase or HEK293 cell extracts. To produce rMrp2R-de, rMrp2R-*was mixed in a 1:1 ratio with
recombinant His-tagged λ-protein phosphatase and incubated for 1 h under rotary stirring at 4 °C. To remove the λ-protein phosphatase, NiNTA beads were added to the sample and further
incubated for 30 min. The sample was spun down to remove the beads with bound λ-protein phosphatase and the supernatant containing the rMrp2R-de was used for further experiments. To generate
the rMrp2R-pho, a HEK293 cell extract was prepared as previously described41; in brief, HEK293 cells were pelleted and washed once in PBS, then resuspended at 30 ×106 cell/mL in HEK293
lysis buffer (50 mM tris/HCl pH 7.5, 10 mM MgCl2, 1 mM EDTA, EDTA-free protease inhibitor tablet, PhosSTOP tablet, 1 mM PMSF) and lysed using a tissue homogenizer. The cell lysate was
centrifuged at 21,000 × _g_ to pellet cell debris and the supernatant was aliquoted in 1 mL and flash frozen in LN2. All the steps were performed at 4 °C or on ice. The HEK293 cell extract
was added during protein purification; after protein elution with elution buffer (50 mM tris/HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 2 mM DTT, 5% Glycerol, 0.02% GDN, 250 mM imidazole) the
protein was exchanged into HEK293 lysis buffer supplemented with 0.02% GDN. 1 mL of HEK293 cell extract and 5 mM ATPwere added to the protein and it was incubated for 30 min at 30 °C. After
incubation, the mixture was re-loaded on the HisTrap column, washed with wash buffer II and eluted with elution buffer. The protein was further purified as in Protein expression and
purification section. MASS SPECTROMETRY ANALYSIS Protein solutions were adjusted to 100 mM triethylammonium bicarbonate (TEAB), reduced with 5 mM tris-2-carboxyethyl phosphine (TCEP),
alkylated with 10 mM iodoacetamide (IAA) and digested overnight with trypsin at a final concentration of 50 ng/μL (Pierce). Peptides were SpeedVac dried and subjected to clean-up with C18
Pierce spin tips followed by phosphopeptide enrichment using the High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo) according to manufacturer’s instructions. Both the eluents
(phosphopeptides) and the flowthroughs were collected for LC-MS analysis. LC-MS analysis was performed on an UltiMate 3000 UHPLC system coupled with the Orbitrap Ascend Mass Spectrometer
(Thermo Scientific). Peptides were loaded onto the Acclaim PepMap 100, 100 μm × 2 cm C18, 5 μm, trapping column at flow rate 10 μL/min and analysed with a nanoEase MZ PST BEH130 1.7 μ 75 μm
× 250 mm C18 capillary column. Mobile phase A was 0.1% formic acid and mobile phase B was 80% acetonitrile, 0.1% formic acid. The separation method was as follows: for 80 min gradient 5%–35%
B, for 5 min up to 95% B, for 5 min isocratic at 95% B, re-equilibration to 5% B in 5 min, for 5 min isocratic at 5% B, at flow rate 300 nL/min. MS scans were acquired in the range of
400–1600 m/z at mass resolution of 120 K, AGC 4 × 105 and max IT 251 ms. Precursors were selected with the top speed mode in 3 sec cycles and isolated for HCD fragmentation with quadrupole
isolation width 0.9 Th. Collision energy was 35% with AGC 1.25 × 105, max IT 200 ms and orbitrap detection at 45 K resolution. Targeted precursors were dynamically excluded for 30 s with 10
ppm mass tolerance. The mass spectra were analysed in Proteome Discoverer 3.0 (Thermo Scientific) with the SequestHT and Comet search engines for peptide identification and quantification.
The precursor and fragment ion mass tolerances were set at 20 ppm and 0.02 Da respectively. Spectra were searched for fully tryptic peptides with maximum 2 missed cleavages. Carbamidomethyl
at C was selected as static modification and oxidation of M, deamidation of N/Q and phosphorylation of S/T/Y were selected as dynamic modifications. Spectra were searched against UniProt
Rattus norvegicus reviewed protein entries, peptide confidence was estimated with the Percolator node and peptides were filtered at q-value < 0.01 based on target-decoy database search.
Phosphorylation localisation probabilities were estimated with the IMP-ptmRS node and peptide quantification was performed with the Minora Feature Detector and Precursor Ions Quantifier
nodes42. The ATPase activity of rMrp2 in different phosphorylation states was measured using the Enzcheck coupled assay kit (Thermofisher). rMrp2 and its phosphorylation variants were
reconstituted into destabilised liposomes consisting of bovine liver total lipid extract (Avanti Polar Lipids). Lipids were rehydrated in a buffer consisting of 20 mM Tris pH 7.5, 150 mM
NaCl at a concentration of 1 mg/ml. The opaque lipid stock was bath sonicated for 10–15 min until the solution turned clear. The small unilamellar vesicles were destabilised by the addition
of 0.2% GDN. rMrp2 was added to a final concentration of 0.1 mg/ml in the lipid mixture to give a molar ratio of rMrp2 to lipid of 1:900 (mol/mol). The ATPase activity of rMrp2 in
destabilised liposomes was measured in the presence of 5 mM ATP and 5 mM MgCl2 at room temperature. For ligand induced ATPase activity assays, probenecid and methotrexate were dissolved in
100% DMSO to make a 100 mM stock solution and were added in the assay to the final concentrations indicated in the figure legends. The reactions were measured using a Labtech CLARIOstar Plus
Microplate Reader. All measurements were performed in triplicate. Analysis and plotting of the data were performed in GraFit 5.0.13. The data were fitted in a 1st order rate equation At =
A∞(1-e-kt) and ATP hydrolysis rate was calculated as product of rate constant and Limit. The statistical significance of experimental data was assessed by one way ANOVA followed by Tukey
test for _p_ < 0.025, _p_ < 0.01 and _p_ < 0.001 as specified in the figure legends. TRANSPORT OF CDF IN PROTEOLIPOSOMES rMrp2 at different phosphorylation states was reconstituted
by detergent removal in a batch-wise procedure; in brief, mixed micelles of detergent, protein and phospholipids were incubated with 0.5 g Amberlite XAD-4 resin under rotatory stirring
(1,200 r.p.m.) at 23 °C for 40 min as previously described43. The composition of the reconstitution was: 15 µL of rMrp2 (75 µg protein) (to avoid saturation of the fluorescent signal in
transport assay, for fully phosphorylated protein only 5 µL i.e. 25 µg proteins were reconstituted), 100 µL of a mixture composed by 60 µL of 10% (w/v) egg yolk phospholipids, containing
7.5% cholesterol (w/w), in the form of sonicated liposomes and 40 µL of 10% C12E8, 70 µL PBS 10X (1× final concentration) in a final volume of 700 µL. The intraliposomal CDF
(5(6)-Carboxy-2′,7′-dichlorofluorescein, Sigma-Aldrich) accumulation was monitored by measuring the fluorescence emission. After reconstitution, 600 µL of proteoliposomes was passed through
a Sephadex G-75 column (0.7 cm diameter ×15 cm height) pre-equilibrated with PBS. Proteoliposomes were added of 5 µM CDF (200 mM stock solution prepared in PBD buffer) and 1 mM probenecid
(100 mM stock prepared in DMSO) as specified in the figure legend. Uptake experiments were started in a 150 µL proteoliposome sample by adding 4 mM ATP together with 4 mM MgCl2 (or
differently as specified in the figure legend), at 25 °C. The transport reaction was stopped after 30 min by passing the mixture through a Sephadex G-75 column (0.6 cm diameter ×8 cm height)
equilibrated with PBS; samples were then, eluted with 1 mL PBS. 0.1% SDS was added to destabilise the proteoliposomes. Fluorometric measurement was performed using the fluorescence
spectrometer LS55 from Perkin Elmer. The fluorescence was measured following time drive acquisition protocol with λ excitation = 504 nm and λ emission = 529 nm (slit 15/10) according to
manufacturer instructions of CDF. Calibration of the fluorescence changes vs CDF concentration has been performed by measuring the fluorescence of known amounts of CDF (from 0 to 6.5 pmoles
in 1 mL) obtaining a linear correlation. The calibration line was used to calculate the pmoles of CDF taken up in rMrp2-harbouring proteoliposomes. A calibration was performed at the end of
each experiment. A liposome control sample i.e. liposomes without reconstituted protein was prepared to quantify the counts given from unspecific lipids fluorescence emission, controls were
subtracted to each sample. The statistical significance of experimental data was assessed by one way ANOVA followed by Tukey test for _p_ < 0.025 as specified in the figure legends.
GENERATION OF IHEPS AND MRP2 ASSAY Human iPSCs (CGT-RCiB10, Cell and Gene Therapy Catapult) were differentiated into iHEPs essentially as described44, except for the Matrigel (Corning)
sandwich and addition of forskolin (5 μM; BioGems) from day 17. On day 19 hepatocyte growth factor was removed and taurine was added (58.4 mg/L; Sigma) and the cells are placed in normoxia.
On day 25 the iHEPs were treated +/−20 nM staurosporine (Generon) for 2 h followed by addition of the MRP2 transport substrate precursor CDFDA (10 µM; Sigma-Aldrich) and NucBlue Live Cell
ReadyProbes Reagent (Thermo Fisher Scientific) and incubated for a further 15 min at 37 °C. CDFDA is cleaved by intracellular esterases to yield fluorescent
5-(and-6)-carboxy-2′,7′–dichlorofluorescein (CDF; green) which is secreted into the bile canaliculi by MRP2. Cell nuclei and accumulation of CDF (green) in bile canaliculi were captured in
images taken on an LSM 880 confocal microscope (ZEISS). Images were analysed using Fiji45. Images were first processed using global thresholding to filter out noise. Then, size-exclusion
filtering was applied to remove particles <5 µm2 to select biologically relevant signals to determine integrated density of CDF staining in the bile canaliculi. The statistical
significance of experimental data was assessed by Student’s test. MRP2 STAINING iHEPs were treated +/−20 nM staurosporine (Generon) for 2 h, after which the cells were fixed in 4% PFA
(Sigma-Aldrich) in PBS for 15 min. The fixed cells were then blocked for 1 h in blocking buffer (0.2% Triton X-100 (Sigma-Aldrich), 3% Donkey-Serum (Sigma-Aldrich) and 1% BSA (Sigma-Aldrich)
in PBS) at room temperature. Next, the cells were stained with a 1:25 dilution of human MRP2 primary antibody (Abcam; ab3373) in blocking buffer overnight at 4 °C. The following day, the
cells were stained with a 1:500 dilution of AlexaFluor 555 donkey anti-mouse antibody (Invitrogen; A-31570) in PBS for 2 h at room temperature, followed by a 5-min incubation with NucBlue
Fixed Cell ReadyProbes Reagent (DAPI; Thermo Fisher Scientific) at room temperature. Cell nuclei (blue) and MRP2 expression at the canalicular membrane (red) were captured in images taken on
an LSM 880 confocal microscope (ZEISS). Images were analysed using Fiji45 as above to determine integrated density of MRP2 staining at the canalicular membrane, total canalicular area,
average canalicular area and canalicular count. The statistical significance of experimental data was assessed by Student’s test. CRYO-EM SAMPLE PREPARATION AND DATA COLLECTION Grids were
prepared with freshly purified protein, rMrp2R-*, at 5 mg/mL. Freshly glow-discharged Quantifoil R1.2/1.3 300-mesh Cu Holey Carbon Grids were treated with 3 mM fluorinated Fos-Choline-8
(Anatrace), followed by the addition of 4 μL of rMrp2 and blotted with filter paper (blot time 3 s, blot force 3), and plunged into liquid ethane using a Vitrobot Mark IV (FEI). The same
procedure was followed for the preparation of the probenecid grids; rMrp2R-* was incubated with 1 mM mM probenecid for 30 min prior to freezing the grids. Cryo-EM data were collected at the
eBIC (Diamond Light Source, UK) on a Titan Krios system (FEI) operated at 300 kV and images were collected using a K3 Imaging detector (Gatan). The data collection was automated, and images
were collected in super-resolution mode Bin2. Data acquisition parameters are summarised in Supplementary Table 1. CRYO-EM DATA PROCESSING AND MODEL BUILDING Movies were imported into
cryoSPARC, corrected for beam-induced motion using patch motion correction and contrast transfer function (CTF) estimations were performed using patch CTF estimation46. Particles were first
picked using a blob picker with minimum diameter of 100 Å and maximum diameter of 200 Å. Particles were inspected, extracted with a 440-pixel box size, and 4.3 million particles were used in
an initial 2D classification with 50 2D classes (with maximum resolution of 6 Å and initial classification uncertainty factor of 2). Seven 2D classes were selected with 1.4 million
particles and further reclassified into 50 2D classes with the same parameters. Eleven 2D classes were selected with 693,363 particles. A sub-batch of 174,000 particles was used to generate
an ab initio model reconstruction. The initial model was used to generate 50 2D templates for a second round of particle picking using cryoSPARC’s template picker, with particle diameter of
190 Å (ab initio was generate only for the nucleotide-free dataset and the generated templates were used to pick particles in both nucleotide-free and probenecid dataset). Particles were
inspected and extracted resulting in a final of 3.8 and 4.9 million particles for rMrp2 nucleotide-free and drug bound, respectively. These particles were downscaled by Fourier Cropping from
440 px box to 110 px box using cryoSPARC’s downsample particles job, then used in 2D classification with 50 classes, maximum resolution of 6 Å, an initial classification uncertainty factor
of 2, force max over poses/shifts set to true, number of online-EM iterations set to 20, and 100 batch size per class. From this, 13 classes for nucleotide-free (1.9 million particles) and 7
classes for probenecid (1 million particles) were selected and submitted to another round of 2D classification with the same parameters. Final number on 8 classes (859,290 particles) were
selected for nucleotide-free and 17 classes (421,743 particles) for probenecid. An initial 3D ab initio with three classes (maximum resolution of 12 Å, initial resolution 35 Å, class
similarity 0.35) resulted in two ‘junk’ classes and one good class for both datasets. The one good class contained 364,687 particles (42.4%) for nucleotide-free and 247,763 particles (58.7%)
for probenecid of the expected size and shape. Non-uniform refinement of the good class was performed using the downscaled particles, then the generated alignments were used to run the
final non uniform refinement47 with the original unbinned particles resulted in a map with GSFSC (0.143) estimated resolution of 3.58 Å and 3.45 Å for the nucleotide-free and drug bound
rMrp2, respectively. Model building and refinement were performed starting from the AlphaFold predicted model for rMrp2 (AF-Q63120-F1). The AF-Q63120-F1 model was split into 5 domains (NBD1
and NBD2, two TMDs and the TMD0), were all individually rigid-body fitted into the map using UCSF ChimeraX48 and real-space refined in phenix49. The model was then subjected to iterative
cycles of refinement and manual rebuilding in COOT50. The refinement statistics are summarised in Supplementary Table 1. MODEL VALIDATION The structural models were validated using
MolProbity (43). Cross-validation to check possibility of overfitting and predictive power of model was performed using half-map validation approach (44). REPORTING SUMMARY Further
information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The cryo-EM maps have been deposited in the Electron
Microscopy Data Bank (EMDB) under accession codes EMD-19431 (nucleotide-free autoinhibited rMrp2) and EMD-19433 (probenecid bound rMrp2). The structural coordinates have been deposited to
the RCSB Protein Data Bank (PDB) under the accession codes 8RQ3 (nucleotide-free autoinhibited rMrp2) and 8RQ4 (probenecid bound rMrp2). Raw data for nucleotide-free autoinhibited rMrp2 and
probenecid bound rMrp2 were submitted to Electron Microscopy Public Image Archive with IDs EMPIAR-11893 and EMPIAR-11894, respectively. The mass spectrometry proteomics data have been
deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD045845. Source data are provided with this paper. REFERENCES * Thomas, C. et al.
Structural and functional diversity calls for a new classification of ABC transporters. _FEBS Lett._ 594, 3767–3775 (2020). Article CAS PubMed PubMed Central Google Scholar *
Jedlitschky, G. et al. ATP-dependent transport of bilirubin glucuronides by the multidrug resistance protein MRP1 and its hepatocyte canalicular isoform MRP2. _Biochem J._ 327, 305–310
(1997). Article CAS PubMed PubMed Central Google Scholar * Paulusma, C. C. et al. A mutation in the human canalicular multispecific organic anion transporter gene causes the
Dubin-Johnson syndrome. _Hepatology_ 25, 1539–1542 (1997). Article CAS PubMed Google Scholar * Kartenbeck, J., Leuschner, U., Mayer, R. & Keppler, D. Absence of the canalicular
isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. _Hepatology_ 23, 1061–1066 (1996). CAS PubMed Google Scholar * Huisman, M. T.,
Chhatta, A. A., van Tellingen, O., Beijnen, J. H. & Schinkel, A. H. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid.
_Int J. Cancer_ 116, 824–829 (2005). Article CAS PubMed Google Scholar * Nies, A. T. & Keppler, D. The apical conjugate efflux pump ABCC2 (MRP2). _Pflug. Arch._ 453, 643–659 (2007).
Article CAS Google Scholar * Hagmann, W., Schubert, J., Konig, J. & Keppler, D. Reconstitution of transport-active multidrug resistance protein 2 (MRP2; ABCC2) in proteoliposomes.
_Biol. Chem._ 383, 1001–1009 (2002). Article CAS PubMed Google Scholar * Cole, S. P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC)
transporter. _J. Biol. Chem._ 289, 30880–30888 (2014). Article CAS PubMed PubMed Central Google Scholar * Leier, I. et al. The MRP gene encodes an ATP-dependent export pump for
leukotriene C4 and structurally related conjugates. _J. Biol. Chem._ 269, 27807–27810 (1994). Article CAS PubMed Google Scholar * Mao, Q., Deeley, R. G. & Cole, S. P. Functional
reconstitution of substrate transport by purified multidrug resistance protein MRP1 (ABCC1) in phospholipid vesicles. _J. Biol. Chem._ 275, 34166–34172 (2000). Article CAS PubMed Google
Scholar * Hagmann, W. et al. Purification of the human apical conjugate export pump MRP2 reconstitution and functional characterization as substrate-stimulated ATPase. _Eur. J. Biochem._
265, 281–289 (1999). Article CAS PubMed Google Scholar * Qu, X. et al. Astragaloside IV enhances cisplatin chemosensitivity in hepatocellular carcinoma by suppressing MRP2. _Eur. J.
Pharm. Sci._ 148, 105325 (2020). Article CAS PubMed Google Scholar * Korita, P. V. et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with
hepatocellular carcinoma. _Oncol. Rep._ 23, 965–972 (2010). CAS PubMed Google Scholar * Stolarczyk, E. I. et al. Casein kinase 2alpha regulates multidrug resistance-associated protein 1
function via phosphorylation of Thr249. _Mol. Pharm._ 82, 488–499 (2012). Article CAS Google Scholar * Chappe, V. et al. Stimulatory and inhibitory protein kinase C consensus sequences
regulate the cystic fibrosis transmembrane conductance regulator. _Proc. Natl Acad. Sci. USA_ 101, 390–395 (2004). Article ADS CAS PubMed Google Scholar * Jia, Y., Mathews, C. J. &
Hanrahan, J. W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. _J. Biol. Chem._ 272,
4978–4984 (1997). Article CAS PubMed Google Scholar * Bakos, E. et al. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. _Mol. Pharm._ 57,
760–768 (2000). Article CAS Google Scholar * Reynolds, E. S., Schlant, R. C., Gonick, H. C. & Dammin, G. J. Fatal massive necrosis of the liver as a manifestation of hypersensitivity
to probenecid. _N. Engl. J. Med._ 256, 592–596 (1957). Article CAS PubMed Google Scholar * Drew, D. et al. GFP-based optimization scheme for the overexpression and purification of
eukaryotic membrane proteins in Saccharomyces cerevisiae. _Nat. Protoc._ 3, 784–798 (2008). Article CAS PubMed PubMed Central Google Scholar * Bakos, E. et al. Functional multidrug
resistance protein (MRP1) lacking the N-terminal transmembrane domain. _J. Biol. Chem._ 273, 32167–32175 (1998). Article CAS PubMed Google Scholar * Westlake, C. J. et al. Identification
of the structural and functional boundaries of the multidrug resistance protein 1 cytoplasmic loop 3. _Biochemistry_ 42, 14099–14113 (2003). Article CAS PubMed Google Scholar * Ford, R.
C. & Beis, K. Learning the ABCs one at a time: structure and mechanism of ABC transporters. _Biochem. Soc. Trans._ 47, 23–36 (2019). Article CAS PubMed Google Scholar * Beuers, U.
et al. Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in
cholestatic rat liver. _Hepatology_ 33, 1206–1216 (2001). Article CAS PubMed Google Scholar * Johnson, Z. L. & Chen, J. Structural Basis of Substrate Recognition by the Multidrug
Resistance Protein MRP1. _Cell_ 168, 1075–1085.e1079 (2017). Article CAS PubMed Google Scholar * Konig, J., Nies, A. T., Cui, Y., Leier, I. & Keppler, D. Conjugate export pumps of
the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. _Biochim. Biophys. Acta_ 1461, 377–394 (1999). Article CAS PubMed
Google Scholar * Liu, F., Zhang, Z., Csanady, L., Gadsby, D. C. & Chen, J. Molecular Structure of the Human CFTR Ion Channel. _Cell_ 169, 85–95.e88 (2017). Article CAS PubMed Google
Scholar * Khandelwal, N. K. et al. The structural basis for regulation of the glutathione transporter Ycf1 by regulatory domain phosphorylation. _Nat. Commun._ 13, 1278 (2022). Article ADS
CAS PubMed PubMed Central Google Scholar * Bickers, S. C., Benlekbir, S., Rubinstein, J. L. & Kanelis, V. Structure of Ycf1p reveals the transmembrane domain TMD0 and the
regulatory region of ABCC transporters. _Proc. Natl Acad. Sci. USA_, 118, https://doi.org/10.1073/pnas.2025853118 (2021). * Zhang, Z., Liu, F. & Chen, J. Molecular structure of the
ATP-bound, phosphorylated human CFTR. _Proc. Natl Acad. Sci. USA_ 115, 12757–12762 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Lundby, A. et al. Quantitative maps of
protein phosphorylation sites across 14 different rat organs and tissues. _Nat. Commun._ 3, 876 (2012). Article ADS PubMed Google Scholar * Trompier, D. et al. Multiple
flavonoid-binding sites within multidrug resistance protein MRP1. _Cell Mol. Life Sci._ 60, 2164–2177 (2003). Article CAS PubMed Google Scholar * Zelcer, N. et al. Evidence for two
interacting ligand binding sites in human multidrug resistance protein 2 (ATP binding cassette C2). _J. Biol. Chem._ 278, 23538–23544 (2003). Article CAS PubMed Google Scholar * Chen, Z.
S. & Tiwari, A. K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. _FEBS J._ 278, 3226–3245 (2011). Article CAS PubMed PubMed Central Google
Scholar * Mor-Cohen, R. et al. Identification and functional analysis of two novel mutations in the multidrug resistance protein 2 gene in Israeli patients with Dubin-Johnson syndrome. _J.
Biol. Chem._ 276, 36923–36930 (2001). Article CAS PubMed Google Scholar * Shapiro, A. B., Fox, K., Lam, P. & Ling, V. Stimulation of P-glycoprotein-mediated drug transport by
prazosin and progesterone. Evidence for a third drug-binding site. _Eur. J. Biochem._ 259, 841–850 (1999). Article CAS PubMed Google Scholar * Yu, Q. et al. Structures of ABCG2 under
turnover conditions reveal a key step in the drug transport mechanism. _Nat. Commun._ 12, 4376 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Ito, K. et al. Mutation of
Trp1254 in the multispecific organic anion transporter, multidrug resistance protein 2 (MRP2) (ABCC2), alters substrate specificity and results in loss of methotrexate transport activity.
_J. Biol. Chem._ 276, 38108–38114 (2001). Article CAS PubMed Google Scholar * Conseil, G., Arama-Chayoth, M., Tsfadia, Y. & Cole, S. P. C. Structure-guided probing of the leukotriene
C(4) binding site in human multidrug resistance protein 1 (MRP1; ABCC1). _FASEB J._ 33, 10692–10704 (2019). Article CAS PubMed Google Scholar * Ito, K., Hoekstra, D. & van
Ijzendoorn, S. C. Cholesterol but not association with detergent resistant membranes is necessary for the transport function of MRP2/ABCC2. _FEBS Lett._ 582, 4153–4157 (2008). Article CAS
PubMed Google Scholar * Kimura, Y., Kodan, A., Matsuo, M. & Ueda, K. Cholesterol fill-in model: mechanism for substrate recognition by ABC proteins. _J. Bioenerg. Biomembr._ 39,
447–452 (2007). Article CAS PubMed Google Scholar * Watson, N. A. et al. Kinase inhibition profiles as a tool to identify kinases for specific phosphorylation sites. _Nat. Commun._ 11,
1684 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Oberoi, J. et al. HSP90-CDC37-PP5 forms a structural platform for kinase dephosphorylation. _Nat. Commun._ 13, 7343
(2022). Article ADS PubMed PubMed Central Google Scholar * Pochini, L., Scalise, M., Galluccio, M., Amelio, L. & Indiveri, C. Reconstitution in liposomes of the functionally active
human OCTN1 (SLC22A4) transporter overexpressed in Escherichia coli. _Biochem. J._ 439, 227–233 (2011). Article CAS PubMed Google Scholar * Blackford, S. J. I. et al. Validation of
Current Good Manufacturing Practice Compliant Human Pluripotent Stem Cell-Derived Hepatocytes for Cell-Based Therapy. _Stem Cells Transl. Med._ 8, 124–137 (2019). Article CAS PubMed
Google Scholar * Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. _Nat. Methods_ 9, 676–682 (2012). Article CAS PubMed Google Scholar * Punjani, A.,
Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. _Nat. Methods_ 14, 290–296 (2017). Article CAS PubMed
Google Scholar * Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. _Nat. Methods_ 17, 1214–1221
(2020). Article CAS PubMed Google Scholar * Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. _Protein Sci._ 30, 70–82 (2021).
Article CAS PubMed Google Scholar * Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. _Acta Crystallogr. D. Struct. Biol._ 74, 531–544 (2018).
Article ADS CAS PubMed PubMed Central Google Scholar * Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. _Acta Crystallogr. D. Biol. Crystallogr._ 60,
2126–2132 (2004). Article ADS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We would like to acknowledge Diamond for access and support of the cryo-EM facilities at the UK
national electron Bio-Imaging Centre (eBIC), proposal BC25127. We would like to thank Prof Erhard Hohenester, Imperial College London, for critical reading of the manuscript. We thank Dr.
Andrew Quigley, Membrane Protein Lab, for access to the CLARIOstar plate reader. T.M. is funded by a fellowship by the project NLHT- Nanoscience Laboratory for Human Technologies, POR
Calabria FESR-FSE 14/20. J.S.C. and T.I.R. are funded by a Cancer Research UK Centre grant (C309/A25144). K.B. is funded by a Medical Research Council grant (MR/N020103/1). AUTHOR
INFORMATION Author notes * These authors contributed equally: Theodoros I. Roumeliotis, Elena Garitta. AUTHORS AND AFFILIATIONS * Department of Life Sciences, Imperial College London, SW7
2AZ, London, UK Tiziano Mazza & Konstantinos Beis * Rutherford Appleton Laboratory, Research Complex at Harwell, Didcot, Oxfordshire, OX11 0FA, UK Tiziano Mazza & Konstantinos Beis *
Department DiBEST (Biologia, Ecologia, Scienze Della Terra) Unit of Biochemistry and Molecular Biotechnology, University of Calabria, 87036, Arcavacata di Rende, Italy Tiziano Mazza &
Cesare Indiveri * Functional Proteomics group, Chester Beatty Laboratories, The Institute of Cancer Research, London, SW3 6JB, UK Theodoros I. Roumeliotis & Jyoti S. Choudhary * Blizard
Institute, Faculty of Medicine and Dentistry, Queen Mary University of London, E1 2A, London, UK Elena Garitta & Kenneth J. Linton * Department of Biochemistry and Biophysics, Stockholm
University, 10691, Stockholm, Sweden David Drew * Department of Metabolism, Digestion & Reproduction, Imperial College London, W12 0NN, London, UK S. Tamir Rashid * CNR Institute of
Biomembranes, Bioenergetics and Molecular Biotechnology (IBIOM), 70126, Bari, Italy Cesare Indiveri Authors * Tiziano Mazza View author publications You can also search for this author
inPubMed Google Scholar * Theodoros I. Roumeliotis View author publications You can also search for this author inPubMed Google Scholar * Elena Garitta View author publications You can also
search for this author inPubMed Google Scholar * David Drew View author publications You can also search for this author inPubMed Google Scholar * S. Tamir Rashid View author publications
You can also search for this author inPubMed Google Scholar * Cesare Indiveri View author publications You can also search for this author inPubMed Google Scholar * Jyoti S. Choudhary View
author publications You can also search for this author inPubMed Google Scholar * Kenneth J. Linton View author publications You can also search for this author inPubMed Google Scholar *
Konstantinos Beis View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS KB designed and managed the overall project. DD provided assistance with
the initial rMrp2 expression and purification. TM performed purification, ATPase assays, transport assays in proteoliposomes, cryo-EM data collection and analysis. TM, CI and KB analysed
transport data. TM and KB built and refined the structures. EG, STR and KJL performed and analysed iHEP transport assays. TIR and JC performed mass spectrometry analysis. KB wrote the
manuscript with help from all the authors. CORRESPONDING AUTHOR Correspondence to Konstantinos Beis. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER
REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Argyris Politis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file
is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Mazza, T., Roumeliotis, T.I., Garitta, E. _et al._ Structural basis for the
modulation of MRP2 activity by phosphorylation and drugs. _Nat Commun_ 15, 1983 (2024). https://doi.org/10.1038/s41467-024-46392-8 Download citation * Received: 05 October 2023 * Accepted:
26 February 2024 * Published: 04 March 2024 * DOI: https://doi.org/10.1038/s41467-024-46392-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative